Guía colombiana para el diagnóstico de la deficiencia de lipasa ácida
DOI:
https://doi.org/10.22516/25007440.180Palabras clave:
Deficiencia de lipasa acida, enfermedad por depósito de esteres de colesterol, enfermedad de Wolman, dislipidemia, hepatomegaliaResumen
Introducción: La deficiencia de lipasa acida lisosomal (LAL-D) es una entidad de herencia autosómica recesiva que lleva a la acumulación de esteres de colesterol y triglicéridos en el hígado, bazo y otros órganos. La edad de inicio y la tasa de progresión son muy variables, lo que posiblemente sea explicado por las mutaciones presentes en el gen LIPA. Las manifestaciones clínicas son las mismas que para otras patologías hepáticas, cardiovasculares y metabólicas, lo que hace difícil reconocerla en la práctica clínica.
Objetivo: Proveer una guía que permita a los clínicos reconocer los principales grupos de riesgo en los cuales se debe sospechar LAL-D y mejorar su diagnóstico.
Metodología: Este documento se diseñó como un Consenso de Expertos en el cual participaron médicos especialistas en Gastroenterología, hepatología, endocrinología, genética, patología y pediatría. Se realizó una revisión de la literatura acerca de las manifestaciones clínicas y de las herramientas para el diagnóstico de LAL-D y se siguió la metodología de técnica de grupo nominal.
Resultados: Se generaron algoritmos diagnósticos por consenso para cada uno de los grupos de riesgo, que facilitaran la sospecha y el diagnóstico de LAL-D.
Conclusiones: Esta guía propone algoritmos para el diagnóstico de LAL-D, basados en el consenso clínico, que buscan optimizar la ruta diagnóstica en los pacientes con esta patología.Descargas
Lenguajes:
esAgencias de apoyo:
Alexion PharmaReferencias bibliográficas
Aslanidis C, Ries S, Fehringer P, Büchler C, Klima H, Schmitz G. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics [Internet]. 1996;33(1):85–93. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8617513
Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: Review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol [Internet]. 2013;58(6):1230–43. Available from: http://dx.doi.org/10.1016/j.jhep.2013.02.014
Sloan HR, Fredrickson DS. Enzyme deficiency in cholesteryl ester storage idisease. J Clin Invest. 1972;51(7):1923–6.
Abramov, A, Schorr, S, Wolman M. Generalized xanthomatosis with calcified adrenals. AMA J Dis Child. 1956;91(3):282–6.
Jones SA, Valayannopoulos V, Schneider E, Eckert S, Banikazemi M, Bialer M, et al. Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants. Genet Med. 2015;(August). Available from: http://www.nature.com/doifinder/10.1038/gim.2015.108
Fredrickson D. Newly recognized disorders of cholesterol metabolism. Ann Intern Med. 1963;58:718.
Fasano T, Pisciotta L, Bocchi L, Guardamagna O, Assandro P, Rabacchi C, et al. Lysosomal lipase deficiency: Molecular characterization of eleven patients with Wolman or cholesteryl ester storage disease. Mol Genet Metab. 2012;105(3):450–6.
Muntoni S, Wiebusch H, Jansen-Rust M, Rust S, Seedorf U, Schulte H, et al. Prevalence of cholesteryl ester storage disease. Arterioscler Thromb Vasc Biol. 2007;27(8):1866–8.
Pisciotta L, Fresa R, Bellocchio A, Pino E, Guido V, Cantafora A, et al. Cholesteryl Ester Storage Disease (CESD) due to novel mutations in the LIPA gene. Mol Genet Metab [Internet]. 2009;97(2):143–8. Available from: http://dx.doi.org/10.1016/j.ymgme.2009.02.007
Grabowski G, Charnas L, Du H. Lysosomal Acid Lipase Deficiencies: the Wolman Disease/Cholesteryl Ester Storage Disease Spectrum. Online Metab Mol Bases Inherit Dis. 2012;1–26.
Freudenberg F, Bufler P, Ensenauer R, Lohse P, Koletzko S. Cholesteryl ester storage disease: An easily missed diagnosis in oligosymptomatic children. Z Gastroenterol. 2013;51(10):1184–7.
Scott SA, Liu B, Nazarenko I, Martis S, Kozlitina J, Yang Y, et al. Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups. Hepatology. 2013;58(3):958–65.
Saito S, Ohno K, Suzuki T, Sakuraba H. Structural bases of Wolman disease and cholesteryl ester storage disease. Mol Genet Metab. 2012;105(2):244–8.
Reiner Ž, Guardamagna O, Nair D, Soran H, Hovingh K, Bertolini S, et al. Lysosomal acid lipase deficiency - An under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235(1):21–30.
Zhang B, Porto AF. Cholesteryl Ester Storage Disease. J Pediatr Gastroenterol Nutr. 2013;56(6):682–5. Available from: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00005176-201306000-00020
Nchimi, A, Rausin, L, Khamis, J. Ultrasound appearance of bowel wall in Wolman’s disease. Pediatr Radiol. 2003;33(4):284–5.
Jones S a., Bernstein D, Bialer M, Dhawan A, Hendriksz C, Whitley CB, et al. Severe and rapid disease course in the natural history of infants with lysosomal acid lipase deficiency. Mol Genet Metab. 2014;111(2):S57–8. Available from: http://www.sciencedirect.com/science/article/pii/S109671921300557X%5Cnhttp://linkinghub.elsevier.com/retrieve/pii/S109671921300557X
Elleder M, Chlumská A, Hyánek J, Poupětová H, Ledvinová J, Maas S, et al. Subclinical course of cholesteryl ester storage disease in an adult with hypercholesterolemia, accelerated atherosclerosis, and liver cancer. J Hepatol. 2000;32(3):528–34.
Elleder M, Ledvinov J, Cieslar P, Kuhn R. Subclinical course of cholesterol ester storage disease (CESD) diagnosed in adulthood - Report on two cases with remarks on the nature of the liver storage process. Virchows Arch A Pathol Anat Histopathol. 1990;416(4):357–65.
Ameis D, Brockmann G, Knoblich R, Merkel M, Ostlund RE, Yang JW, et al. A 5’ splice-region mutation and a dinucleotide deletion in the lysosomal acid lipase gene in two patients with cholesteryl ester storage disease. J Lipid Res. 1995;36(2):241–50.
Riva S, Spada M, Sciveres M, Minervini M, Cintorino D, Maggiore G, et al. Hepatocarcinoma in a child with cholesterol ester storage disease. Dig Liver Dis. 2008;40(9):784.
Drebber U, Andersen M, Kasper HU, Lohse P, Stolte M, Dienes HP. Severe chronic diarrhea and weight loss in cholesteryl ester storage disease: a case report. World J Gastroenterol. 2005;11(15):2364–6.
Hamilton J, Jones I, Srivastava R, Galloway P. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta [Internet]. 2012;413(15–16):1207–10. Available from: http://dx.doi.org/10.1016/j.cca.2012.03.019
Civallero G, Michelin K, de Mari J, Viapiana M, Burin M, Coelho JC, et al. Twelve different enzyme assays on dried-blood filter paper samples for detection of patients with selected inherited lysosomal storage diseases. Clin Chim Acta. 2006;372(1–2):98–102.
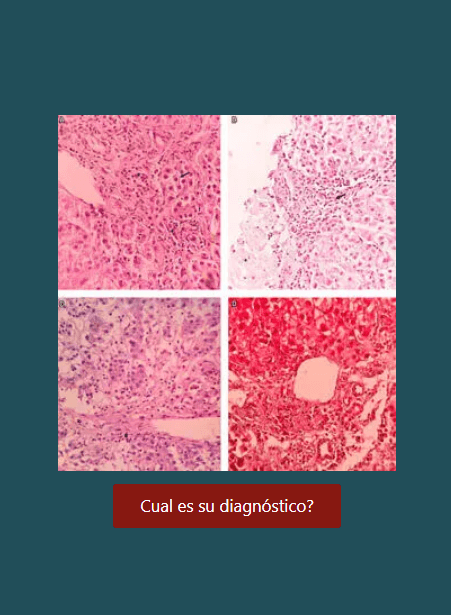
Húlková H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology. 2012;60(7):1107–13.
Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, et al. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142(7):1592–609. Available from: http://dx.doi.org/10.1053/j.gastro.2012.04.001
Wolf a D, Lavine JE. Hepatomegaly in neonates and children. Pediatr Rev. 2000;21(9):303–10.
Linari S, Castaman G. Clinical manifestations and management of Gaucher disease. Clin Cases Miner Bone Metab [Internet]. 2015;12(2):157–64. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4625773&tool=pmcentrez&rendertype=abstract
Dahl S Vom, Harzer K, Rolfs A, Albrecht B, Niederau C, Vogt C, et al. Hepatosplenomegalic lipidosis: What unless Gaucher? Adult cholesteryl ester storage disease (CESD) with anemia, mesenteric lipodystrophy, increased plasma chitotriosidase activity and a homozygous lysosomal acid lipase -1 exon 8 splice junction mutation. J Hepatol. 1999;31(4):741–6.
Masi L, Brandi ML. Gaucher disease: the role of the specialist on metabolic bone diseases. Clin Cases Miner Bone Metab. 2015;12(2):165–9.
Zampieri S, Filocamo M, Pianta A, Lualdi S, Gort L, Coll MJ, et al. SMPD1 Mutation Update: Database and Comprehensive Analysis of Published and Novel Variants. Hum Mutat. 2015;
Rhein C, Mühle C, Kornhuber J, Reichel M. Alleged Detrimental Mutations in the SMPD1 Gene in Patients with Niemann-Pick Disease. Int J Mol Sci. 2015;16(6):13649–52. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4490514&tool=pmcentrez&rendertype=abstract
McGovern MM, Wasserstein MP, Giugliani R, Bembi B, Vanier MT, Mengel E, et al. A prospective, cross-sectional survey study of the natural history of Niemann-Pick disease type B. Pediatrics. 2008;122(2):e341–9.
Bodamer O a, Feillet F, Lane RE, Lee PJ, Dixon M a, Halliday D, et al. Utilization of cornstarch in glycogen storage disease type Ia. Eur J Gastroenterol Hepatol [Internet]. 2002;14(11):1251–6. Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id= 12439121&retmode=ref&cmd=prlinks%5Cnpapers3://publication/uuid/CB712C8E-D63E-4AC0-9B42-4C5DF0A2230C
Avaria, M, Kleinsteuber S. Trastorno del metabolismo de hidratos de carbono. In: Editores R, editor. Errores innatos en el metabolismo del niño. 3ra ed. 2010. p. 458–66.
Grundy SM, Brewer HB, Cleeman JI, Smith SC, Lenfant C. Definition of Metabolic Syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on Scientific Issues Related to Definition. Circulation. 2004;109(3):433–8.
Alberti KGMM, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the Metabolic Syndrome: A Joint Interim Statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International . Circulation. 2009;120(16):1640–5. Available from: http://circ.ahajournals.org/content/120/16/1640.abstract%5Cnhttp://circ.ahajournals.org/content/120/16/1640.full.pdf
Weiss R, Bremer AA, Lustig RH. What is metabolic syndrome, and why are children getting it? Ann N Y Acad Sci. 2013;1281(1):123–40.
Varghese M. Familial hypercholesterolemia: A review. Ann Pediatr Cardiol [Internet]. 2014;7(2):107. Available from: http://www.annalspc.com/text.asp?2014/7/2/107/132478
Wiegman A, Gidding SS, Watts GF, Chapman MJ, Ginsberg HN, Cuchel M, et al. Familial hypercholesterol??mia in children and adolescents: Gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015;36(36):2425–37.
Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease. Eur Heart J. 2013;34(45):3478–90.
Mata P, Alonso R, Ruíz-García A, et al. Hiperlipidemia familiar combinada: documento de consenso. Aten Primaria. 2014;46(8):440–6. Available from: http://dx.doi.org/10.1016/j.aprim.2014.04.013

Descargas
Publicado
Cómo citar
Número
Sección
Licencia
Aquellos autores/as que tengan publicaciones con esta revista, aceptan los términos siguientes:
Los autores/as ceden sus derechos de autor y garantizarán a la revista el derecho de primera publicación de su obra, el cuál estará simultáneamente sujeto a la Licencia de reconocimiento de Creative Commons que permite a terceros compartir la obra siempre que se indique su autor y su primera publicación en esta revista.
Los contenidos están protegidos bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional.

| Estadísticas de artículo | |
|---|---|
| Vistas de resúmenes | |
| Vistas de PDF | |
| Descargas de PDF | |
| Vistas de HTML | |
| Otras vistas | |